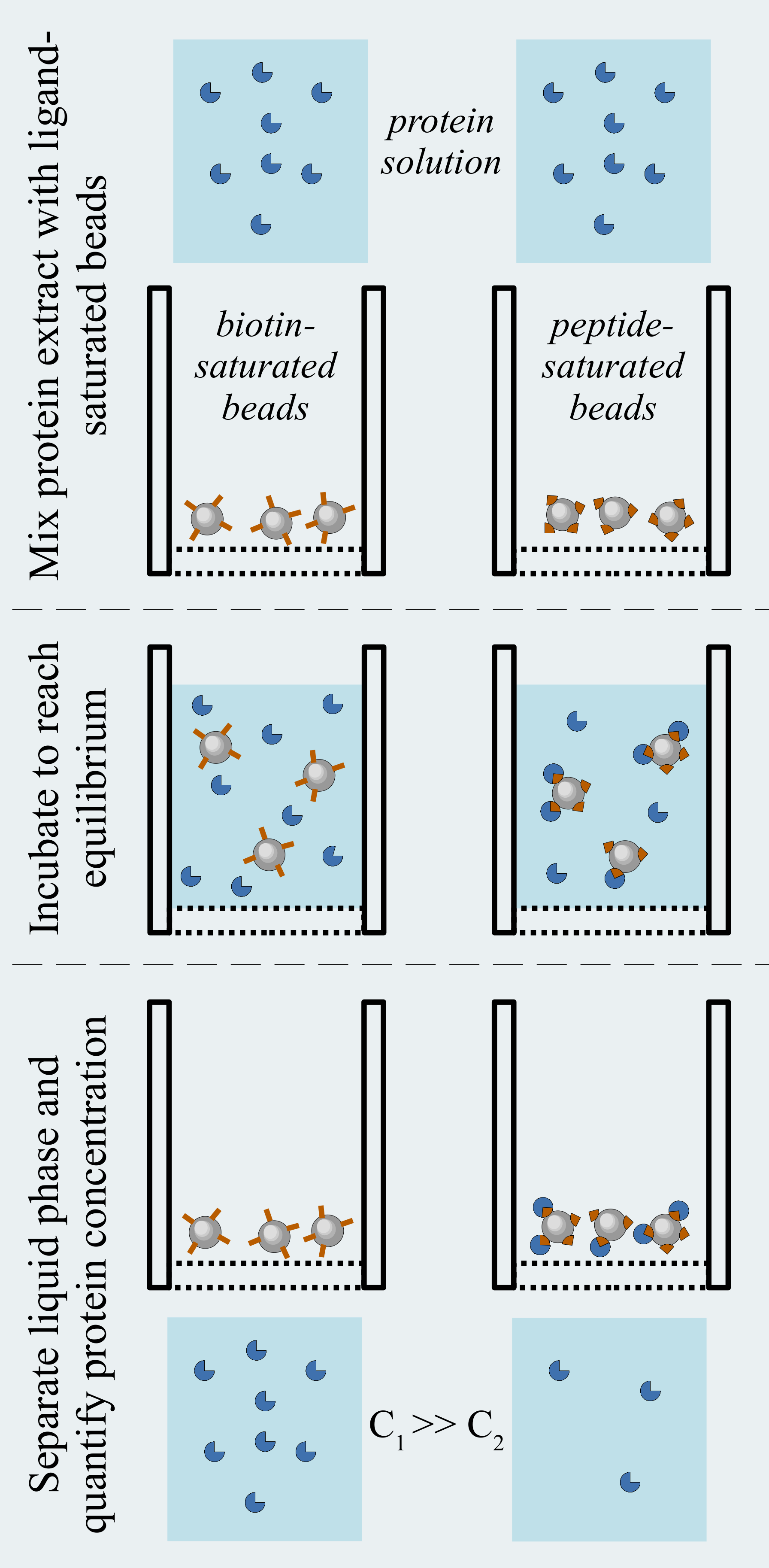
PDZ-PBM interactions are relatively weak, with dissociation constants ranging from submicromolar to millimolar. This makes the task of quantifying thousands of interactions challenging. For this reason, our consortium developed a simple chromatographic approach, called the "holdup assay", that can reliably quantify weak domain-motif interactions at high throughput.
The holdup assay evaluates the concentrations of free (unbound) proteins in conditions approaching thermodynamic equilibrium. One partner of the bi-molecular interaction is immobilized on a solid resin and is mixed with a known amount of the other partner. When the equilibrium is reached, the solid phase is quickly separated from the liquid fraction, and the depleted concentration of the soluble partner is determined. The stronger the depletion, the stronger the bi-molecular interaction. Several phenomena may occur leading to improper depletion estimate:
(1) Some proteins can be retained even on an inert resin, which would cause a false positive binding. To minimize the consequence of this effect, we use a negative control, where the resin does not contain the putative partner protein. Considering the ratio of supernatant concentrations obtained from the binding experiment and the negative control, one can determine the binding intensity "BI", which is the real depleted fraction of the studied protein. For example, a BI=1.0 means that the entire amount of the protein is captured by the resin bearing the putative partner, as compared to the negative control resin. A BI=0.0 means that no detectable enrichment was observed on the resin bearing the putative partner.
(2) Internal standards are employed to eliminate effects of volume variation from one measurement to another. An internal standard is a detectable molecule (protein, fluorescent molecule...) that is added to the protein solution, and is not expected to bind to the peptide immobilized on the resin.
(3) Any leakage of captured proteins from the resin can cause imprecise results. To avoid these, we use streptavidin beads that capture biotinylated peptides. The resin is first incubated with an excess amount of biotinylated peptide (or biotin) to achieve saturation. Then, after a quick washing step, a large amount of biotin solution is added to the resin to deplete all unligated streptavidin (if still present). The extremely slow dissociation rate of the streptavidin-biotin interaction allows us to re-use the same peptide-loaded resin several times, using an appropriate washing step between each measurement.
(4) If the separation of the liquid phase is slow, the equilibrium can be imbalanced, leading to an apparently increased depletion factor. For this step, we are using fast filtration with filter plates and fast centrifugation. According to our benchmarks based on orthogonal affinity measurements, this phenomenon does not occur, or only has a marginal effect on our data.
Our PDZ library is expressed as his6-MBP fusion proteins. The PDZ library that is used for the holdup assay can originate from various sources. The simplest sample that we can use to measure holdup is a total bacterial extract, that is diluted to achieve a uniform concentration for the different PDZ samples (usually 4 µM). In this case, the library is complemented with lysozyme as an internal standard and the concentration measurement is performed on a microcapillary electrophoresis instrument in order to identify and quantify the protein of interest. While this assay provides a very reliable affinity measurement, it is both expensive and labour intensive. These measurements are indicated as LYSC in the database (lysate with caliper readout).
Alternatively, the overexpressed PDZ library can be purified on Ni-beads. This results in a rather pure library, although some contaminants and degradations can still be present. This library is mixed with fluorescein as an internal standard and the concentration measurement is based on the intrinsic Trp-fluorescence of the PDZ-proteins. This holdup assay is both fast and cheap and is perfect to quickly explore the interactome. However, the resulting affinity data may be somewhat less reliable because of the low specificity of the fluorescent readout and the lower sample quality as compared to the other assays. These measurements are indicated as SAPF in the database (single affinity purified with fluorescent readout).
Finally, a large portion of the human PDZome was also purified in large amounts using a double affinity strategy. Bacterial extracts were first purified using Ni-resin and the captured MBP-fused PDZ domains were further purified on amylose columns. This is our most pure library that is complemented with fluorescein and mCherry as internal standards before the holdup measurements. Concentration measurement is performed using the intrinsic Trp-fluorescence of the PDZ-proteins. These measurements are indicated as DAPF in the database (double affinity purified with fluorescent readout).
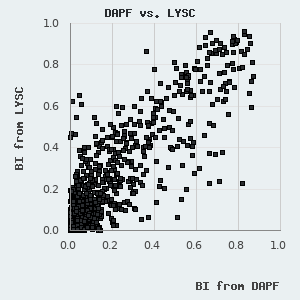
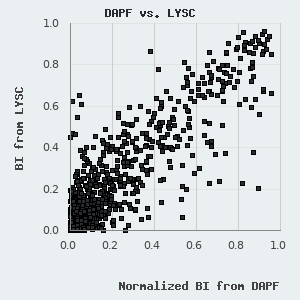
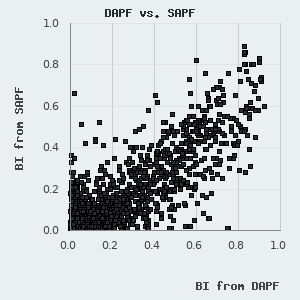
We have found that the BI values determined by LYSC and DAPF measurements show remarkable correlation, although the fluorescent readout provides somewhat smaller BI values (see figure on the left). This is most likely due to the presence of the non-specific nature of the fluorescent concentration measurement and is caused by minor protein contaminants or protein degradation products that may cumulatively produce a significant Trp-fluorescence signal, which will not be selectively retained by the target peptide immobilized on the resin. We use the unbiased BI values from LYSC measurements to figure out an empirical correction factor that is applied to normalize for PDZ degradation/contamination. This is possible since we assay multiple peptides against the same purified sample in DAPF experiments. Unfortunately, due to the experimental layout (1 motif versus 266 domains), no corrections could be applied in (certain) SAPF experiments.
The binding intensity of a holdup reaction is reflective of the chemical equilibrium and is defined by three parameters: the dissociation constant and the total concentration of the two reactants. During the assay preparation, we set the concentration of the soluble partner (the PDZ-protein) to a known value, therefore the binding intensity is related to only two unknown parameters. We can also assume that the concentration of the immobilized peptide is the same for all holdup experiments that were performed with the same peptide. To estimate this concentration, we measure the dissociation constants of several PDZ-PBM interactions using competitive fluorescence polarization (FP). By combining the known PDZ domain concentration, with the measured dissociation constants, and the measured binding intensity, one can easily calculate the concentration of the peptide ligand. This calculation can be performed using several BI-Kd pairs in order to deduce an average peptide concentration that can be used to convert the entire BI profile (all holdup measurements) of the same peptide.
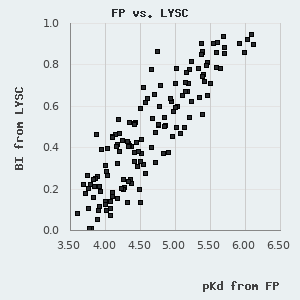
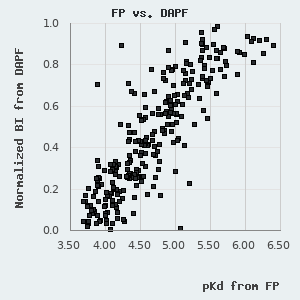
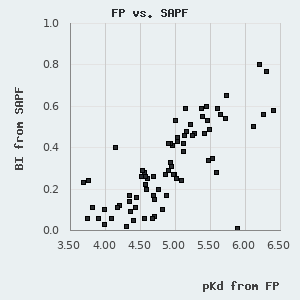
On this database, we show a composite affinity, based on the three types of holdup experiments by prioritizing DAPF, over LYSC and SAPF measurements. For practical reasons, we show pKd, i.e. the negative 10-base logarithm of the dissociation constant (expressed in Molar unit), a value that is proportional to the change in Gibbs free energy during the binding event. pKd can be easily converted to the standard change in Gibbs free energy by applying the following formula:
ΔG0=pKd/2.303RT
where R is the gas constant and T is the temperature in K. All experiments were performed at room temperature.
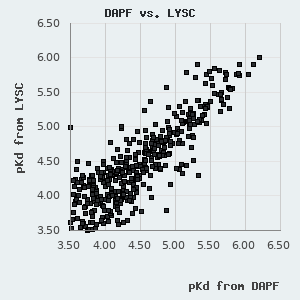
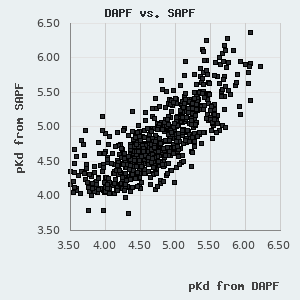
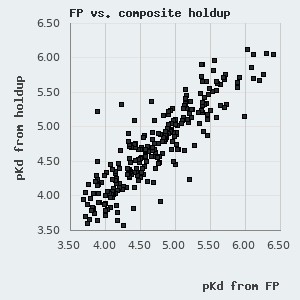
One can find a normal distribution between the orthogonal pKd values derived from FP and holdup measurements with a standard deviation of 0.227 pKd. By assuming that neither of these methods diverge from real affinities systematically, we can use this parameter to describe the robustness of the affinities presented in this database. One can also use it to estimate the confidence of an affinity difference using the following empirical rule: >0.227 pKd difference indicates ~68.3% confidence (1 σ), >0.454 pKd difference indicates ~95.5% confidence (2 σ), and >0.681 pKd difference indicates a 99.7% confidence (3 σ).
- Capturing protein-protein complexes at equilibrium: the holdup comparative chromatographic retention assay. S. Charbonnier et al. Protein Expr Purif., 2006.
- Quantifying domain-ligand affinities and specificities by high-throughput holdup assay. R Vincentelli et al. Nature methods, 2015
- Benchtop holdup assay for quantitative affinity-based analysis of sequence determinants of protein-motif interactions. A Bonhoure et al. Analytical Biochemistry, 2020
- Dual specificity PDZ-and 14-3-3-binding motifs: a structural and interactomics study. G Gogl et al. Structure, 2020