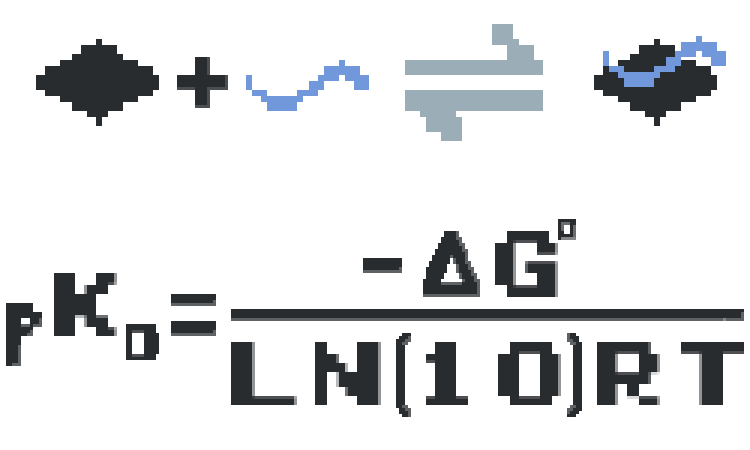
ProfAff (standing for Profiling Affinities) is a proof of concept interactomic database that not only stores lists of interactions found between macromolecules, but rather directly present measured biophysical properties. This is in clear contrast compared to conventional interactomic databases, such as IntAct or BioGrid, where every interaction is considered as equal and detection thresholds or units of measurements are not reported directly. At the moment, ProfAff aims to collect equilibrium dissociation constants (in the form of negative logarithm values, reffered to as pKd). These binding parameters are intrinsic biophysical constant values that describe the binding properties of interactions and also determine amounts of complex formations in a given cellular contexts under binding equilibrium. We are welcoming any quantitatative quantitative interactomic dataset (e.g. binding constants) that we could upload in future releases of ProfAff. We are also open to include other types of biophysical parameters that are currently not stored in the current version of the database, such as kinetic or thermodynamic parameters.
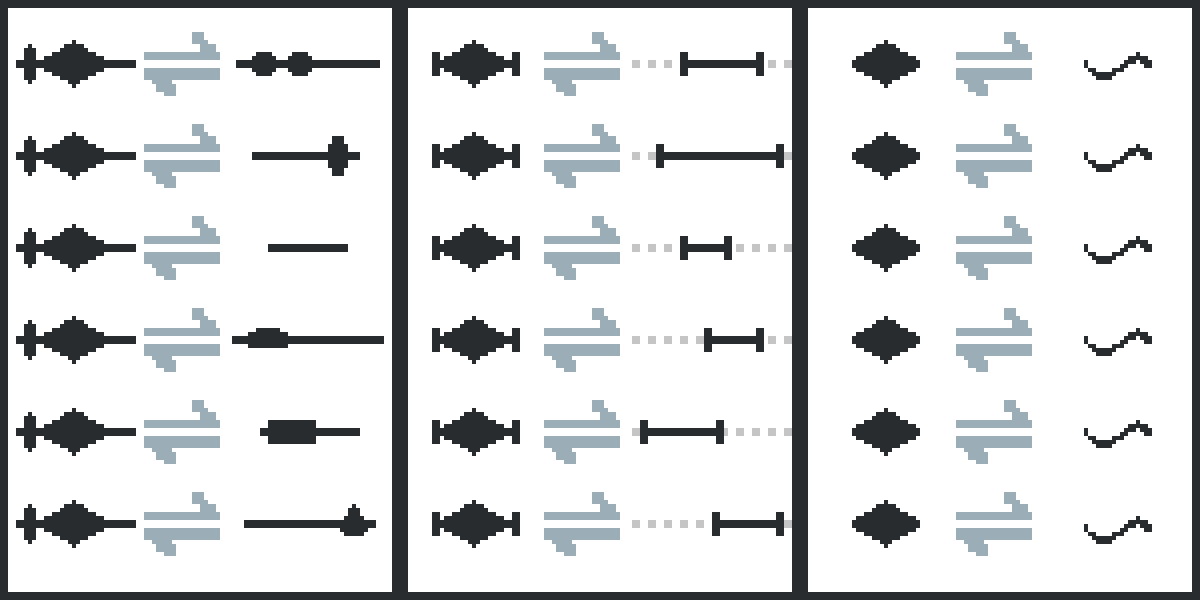
The quantitative interactome is organized on three levels on ProfAff: molecules, fragments, and sites.
Molecules: For a first approximation, all measurements done between the same type of molecules are combined. (For example, any affinities measured between two proteins are combined and the average affinities are reported. The only exception from this averaging occurs when a particular interaction was measured with a given protein variant.) These affinities can be misleading, since this averaging procedure can combine the measured affinities of unrelated fragments and can hide cooperative effects, or can apparently decrease the reliability of the reported affinities. Example: SCRIB, where affinities measured using various fragments covering different functional sites are combined per interactin molecule.

Fragments: This interactomic profile contains the least amount of dimension reduction as only the results of measurements performed between the exact same molecule fragments are combined. Example: 725-735 fragment of RPS6KA1, where affinities are only combined when the experiment was performed using the exact same binding fragments. Here, the selected fragment has multiple variants and their affinities are listed separately.

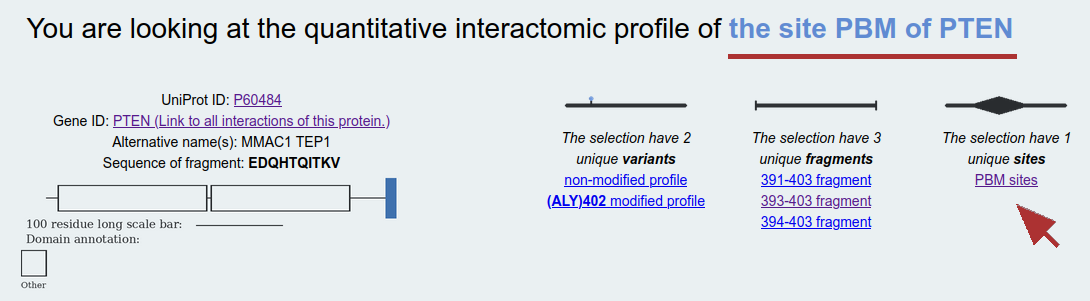
Sites: The results of some experiments done with slighlty different fragments are functionally redundant. For example, when the binding properties of a small interactiong peptide with different number of flanking residues can have very similar affinities. On this layer, we combine site-specific affinties, regardless of the used fragments. In case the selected site interacts with SLiMs following a specific consensus motif, affinity-weighted consensus LOGOs are also calculated automatically. Example: PBM site of PTEN, where affinities measured using slightly different fragments covering the same functional site are combined. Here, the selected site have multiple variants and their affinities are listed separately.